
Tekst oparty na pracy
The Adipokine Hypothesis of Heart Failure With a Preserved Ejection Fraction: A Novel Framework to Explain Pathogenesis and Guide Treatment
Podsumowanie:
Przez wiele lat otyłość była postrzegana wyłącznie jako jeden z wielu czynników ryzyka chorób serca. Najnowsze badania pokazują jednak coś znacznie ważniejszego: otyłość – zwłaszcza trzewna (brzuszna) – może być kluczowym mechanizmem prowadzącym do niewydolności serca z zachowaną frakcją wyrzutową (HFpEF). Tkanka tłuszczowa – aktywny narząd, nie „magazyn energii” Tkanka tłuszczowa nie jest bierna. To największy narząd wydzielniczy organizmu, produkujący setki biologicznie aktywnych substancji – tzw. adipokin. U osób szczupłych adipokiny pełnią funkcje ochronne:
-
wspierają prawidłową pracę serca,
-
ograniczają stan zapalny,
-
poprawiają funkcję naczyń i metabolizm.
W przypadku otyłości trzewnej sytuacja radykalnie się zmienia. Przerośnięta i zapalnie aktywna tkanka tłuszczowa zaczyna wydzielać zupełnie inny zestaw sygnałów biologicznych, które:
-
nasilają stan zapalny w całym organizmie,
-
sprzyjają retencji sodu i wzrostowi objętości krwi,
-
prowadzą do przerostu i włóknienia mięśnia sercowego,
-
zaburzają mikrokrążenie wieńcowe.
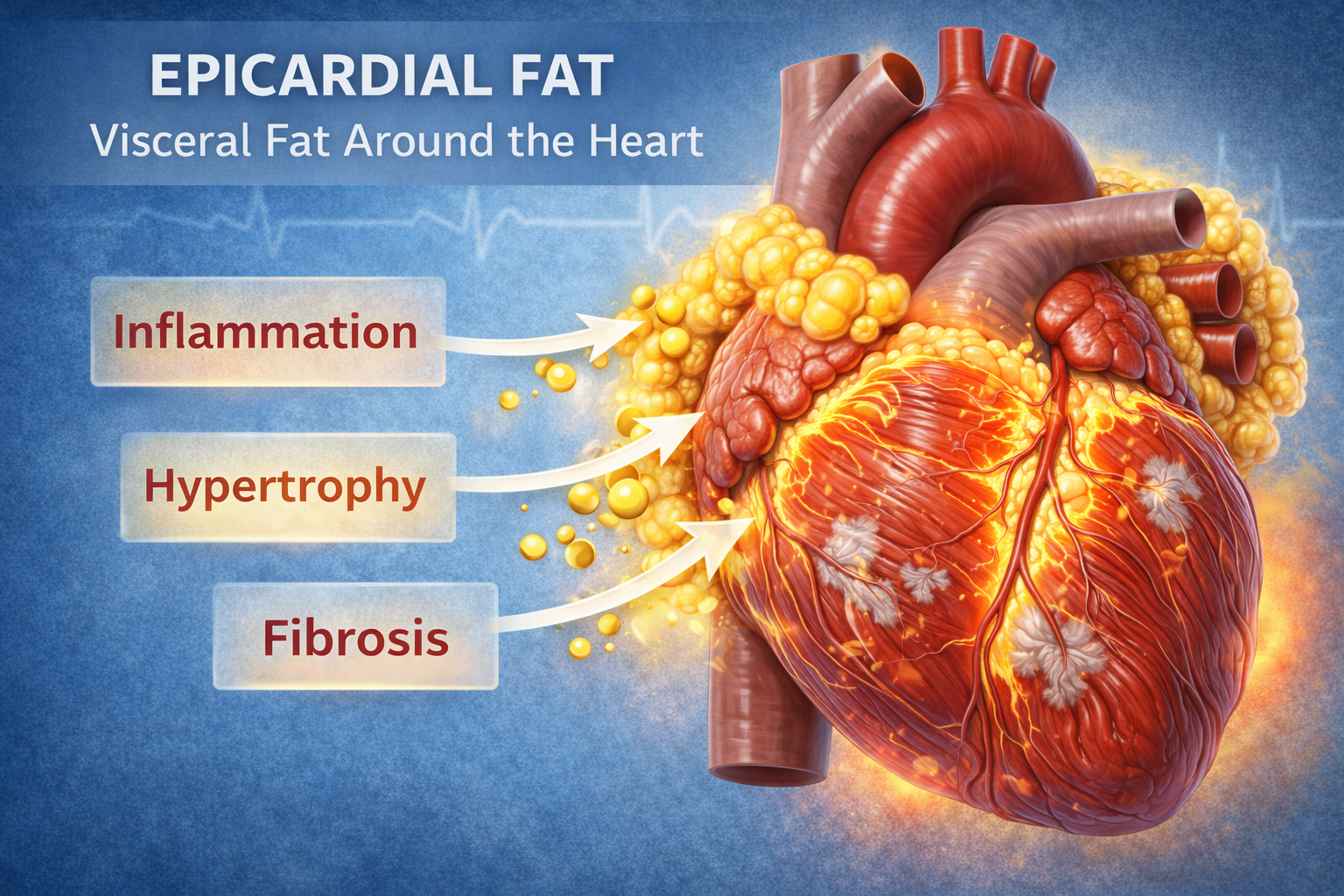
Dlaczego serce jest szczególnie narażone?Serce jest wyjątkowo wrażliwe na działanie adipokin z dwóch powodów.
Po pierwsze – krążące adipokiny docierają do niego z krwią.
Po drugie – serce jest dosłownie otoczone tkanką tłuszczową nasierdziową, która w otyłości staje się silnie prozapalna i oddziałuje na mięsień sercowy bezpośrednio, „z bliska”. To właśnie ten mechanizm sprzyja rozwojowi:
-
sztywności lewej komory,
-
zaburzeń napełniania rozkurczowego,
-
nietolerancji wysiłku,
czyli kluczowych cech HFpEF, nawet przy prawidłowej frakcji wyrzutowej.
HFpEF – choroba metaboliczno-zapalna HFpEF nie jest jedynie „łagodniejszą formą” niewydolności serca. Coraz więcej danych wskazuje, że jest to choroba metaboliczno-zapalna, ściśle związana z:
-
otyłością trzewną,
-
insulinoopornością,
-
nadciśnieniem,
-
cukrzycą typu 2.
To tłumaczy, dlaczego u większości pacjentów z HFpEF współistnieje otyłość brzuszna, a klasyczne leczenie kardiologiczne bywa niewystarczające. Dlaczego nowoczesne leki działają? Leki, które obecnie wykazują skuteczność w HFpEF – takie jak flozyny, antagoniści aldosteronu, sakubitryl/walsartan czy agoniści receptorów GLP-1 lub GLP-1/GIP - nie działają wyłącznie na serce i nerki. One również:
-
zmniejszają masę tłuszczu trzewnego,
-
poprawiają biologię tkanki tłuszczowej,
-
przywracają korzystny profil adipokin,
-
redukują stan zapalny i włóknienie.
Co istotne, korzyści te często są nieproporcjonalnie większe niż sama utrata masy ciała, co podkreśla, że kluczowa jest jakość tkanki tłuszczowej, a nie tylko liczba kilogramów. Nowy paradygmat leczenia Z tego punktu widzenia HFpEF można postrzegać jako chorobę, która:
-
zaczyna się w tkance tłuszczowej,
-
rozwija się poprzez zaburzenia sygnałów metabolicznych i zapalnych,
-
prowadzi do strukturalnych i czynnościowych zmian w sercu.
Dlatego skuteczne leczenie HFpEF powinno być kompleksowe i obejmować:
-
redukcję tłuszczu trzewnego,
-
normalizację funkcji tkanki tłuszczowej,
-
leczenie metaboliczne,
-
a nie wyłącznie klasyczne podejście „kardiocentryczne”.
Wstęp
Niewydolność serca z zachowaną frakcją wyrzutową (HFpEF) stanowi obecnie około 50–60% wszystkich przypadków niewydolności serca(ponad 26 mln osób na całym świecie, około 1 mln w Polsce), a jej częstość występowania systematycznie rośnie. Wzrost ten jest ściśle związany ze starzeniem się populacji oraz narastającą epidemią otyłości, cukrzycy typu 2 i nadciśnienia tętniczego. W przeciwieństwie do HFrEF, zapadalność na HFpEF zwiększa się przede wszystkim u osób w wieku podeszłym i kobiet. Rokowanie w HFpEF pozostaje niekorzystne i porównywalne z HFrEF,(niewydolność serca z obniżoną frakcją wyrzucania) z wysoką częstością hospitalizacji i istotną śmiertelnością długoterminową. Pomimo rosnącego obciążenia epidemiologicznego, skuteczne leczenie przyczynowe HFpEF nadal pozostaje ograniczone, co podkreśla potrzebę lepszego zrozumienia mechanizmów patofizjologicznych leżących u podstaw tej choroby.
Niewydolność serca z zachowaną frakcją wyrzutową (HFpEF) pozostaje jednym z największych wyzwań współczesnej kardiologii. Przez lata była postrzegana jako heterogenny zespół kliniczny wynikający z nakładania się licznych chorób współistniejących, takich jak nadciśnienie tętnicze, cukrzyca typu 2, otyłość, migotanie przedsionków, przewlekła choroba nerek czy stan zapalny układowy. Takie podejście, choć powszechne, nie doprowadziło ani do powstania spójnego modelu patogenetycznego, ani do skutecznych terapii modyfikujących przebieg choroby. Coraz więcej danych wskazuje jednak, że współwystępowanie tych zaburzeń nie jest przypadkowe, lecz odzwierciedla istnienie wspólnego mechanizmu biologicznego. W tym kontekście szczególną rolę przypisuje się otyłości trzewnej – nadmiernej i dysfunkcyjnej tkance tłuszczowej zlokalizowanej wokół narządów wewnętrznych. Tkanka ta pełni aktywną funkcję endokrynną i immunologiczną, wydzielając adipokiny oraz cytokiny prozapalne o istotnym znaczeniu kardiometabolicznym.
Hipoteza adipokinowa HFpEF zakłada, że dysfunkcyjna tkanka tłuszczowa trzewna stanowi centralny czynnik patogenetyczny choroby. Poprzez indukcję insulinooporności, przewlekłego stanu zapalnego, aktywację układów neurohormonalnych oraz bezpośredni wpływ na mięsień sercowy, naczynia i nerki, otyłość trzewna prowadzi do przerostu i sztywności lewej komory, zwłóknienia przedsionków, dysfunkcji mikrokrążenia i zaburzeń objętościowych-kluczowych cech HFpEF. Starzejące się serce wykazuje szczególną podatność na te niekorzystne bodźce biologiczne. W takim znaczeniu otyłość trzewna nie jedną z wielu chorób współistniejących, lecz potencjalnym wspólnym mianownikiem patofizjologicznym HFpEF, integrującym obserwacje epidemiologiczne, metaboliczne i kardiologiczne w jeden spójny model choroby. HFpEF wykazuje wyraźną predylekcję do występowania u kobiet, u których obserwuje się większy wzrost ciśnienia napełniania lewej komory przy zwiększeniu objętości krwi oraz większą sztywność serca i tętnic niż u mężczyzn. Kobiety mają również relatywnie większy udział tkanki tłuszczowej w masie ciała i są szczególnie predysponowane do rozrostu tłuszczu nasierdziowego oraz zaburzeń sygnalizacji adipokinowej, co wspiera hipotezę adipokinową HFpEF. Choć otyłość klinicznie definiuje się za pomocą BMI, kluczowe znaczenie patogenetyczne ma nadmiar i dysfunkcja tkanki tłuszczowej trzewnej. Dlatego hipoteza adipokinowa dotyczy także osób bez „otyłości” według BMI, u których obecny jest nadmiar tłuszczu trzewnego gromadzącego się wokół narządów i w jamie brzusznej(zespół TOFI-szczupły na zewnątrz, otyły wewnątrz). Z tego powodu lepszym markerem otyłości trzewnej lub centralnej jest współczynnik tali/wzrost z progiem odcięcia 0,5.
Podobieństwa biologiczne i kliniczne otyłości oraz HFpEF
Otyłość, zwłaszcza trzewna, i HFpEF wykazują istotne podobieństwa w badaniach epidemiologicznych i klinicznych. Oba stany charakteryzują się zbliżonymi zmianami strukturalnymi i czynnościowymi serca, podobnymi profilami neurohormonalnymi i prozapalnymi oraz wspólnymi cechami molekularnego stresu kardiomiocytów. Tak wyraźne nakładanie się cech otyłości i HFpEF sugeruje istnienie wspólnego mechanizmu patogenetycznego, a nie przypadkowego współwystępowania dwóch chorób. Zbieżność ta wspiera koncepcję, że HFpEF może być jedną z manifestacji zaburzeń wywołanych przez dysfunkcyjną tkankę tłuszczową trzewną i jej sygnalizację adipokinową.
Nieprawidłowości hemodynamiczne, neurohormonalne i kardiologiczne w otyłości
Otyłość, szczególnie trzewna, prowadzi do zaburzeń regulacji objętości krwi i ciśnienia tętniczego poprzez nasiloną retencję sodu w nerkach. Wynika ona z aktywacji układu współczulnego oraz osi renina–angiotensyna–aldosteron, z istotnym udziałem adipokin, zwłaszcza leptyny, która stymuluje wydzielanie aldosteronu i aktywację receptorów mineralokortykoidowych. Dodatkowo otyłość osłabia działanie peptydów natriuretycznych, co sprzyja nadciśnieniu zależnemu od objętości.
Zaburzenia neurohormonalne bezpośrednio wpływają na serce, aktywując procesy przerostu i włóknienia mięśnia sercowego oraz zwiększając sztywność naczyń. Przewlekły, niskiego stopnia stan zapalny i dysfunkcja mikrokrążenia wieńcowego mogą występować jeszcze przed rozwojem jawnej choroby sercowo-naczyniowej. Na poziomie komórkowym otyłość wiąże się ze stresem oksydacyjnym, dysfunkcją mitochondriów i uszkodzeniem kardiomiocytów, czego wykładnikiem są podwyższone stężenia troponin sercowych. Klinicznie prowadzi to do upośledzenia rozkurczu lewej komory, wzrostu ciśnienia napełniania i powiększenia lewego przedsionka, nawet przed spełnieniem kryteriów HFpEF. Równolegle dochodzi do hiperfiltracji kłębuszkowej i przebudowy nerek, co sprzyja rozwojowi przewlekłej choroby nerek i dodatkowo napędza mechanizmy prowadzące do HFpEF.
Nieprawidłowości hemodynamiczne i neurohormonalne w HFpEF
Mechanizmy patofizjologiczne charakterystyczne dla otyłości, zwłaszcza trzewnej, są również nasilone u pacjentów z HFpEF. Do podstawowych cech HFpEF należą retencja sodu, zwiększona objętość osocza oraz nadciśnienie tętnicze. U większości chorych występuje upośledzone wydalanie soli przez nerki oraz przewlekła aktywacja układu współczulnego, prowadząca do zmniejszenia pojemności żylnej i wzrostu ciśnienia napełniania lewej komory, szczególnie w warunkach stresu lub wysiłku. HFpEF wiąże się z nasilonym zaburzeniem osi neurohormonalnych: podwyższonym stężeniem aldosteronu i leptyny oraz obniżoną skutecznością peptydów natriuretycznych, częściowo na skutek zwiększonej aktywności neprylizyny. Zaburzenia te mają znaczenie prognostyczne, sprzyjają nadciśnieniu zależnemu od objętości i odpowiadają na leczenie antagonistami receptora mineralokortykoidowego, inhibitorami neprylizyny oraz lekami opartymi na inkretynach. Nieprawidłowa sygnalizacja neurohormonalna w HFpEF prowadzi do przerostu i włóknienia mięśnia sercowego, wzrostu sztywności komór oraz dysfunkcji mikrokrążenia wieńcowego. Przewlekły stan zapalny i stres oksydacyjny skutkują uszkodzeniem kardiomiocytów, dysfunkcją mitochondriów oraz zaburzeniami gospodarki wapniowej, czego klinicznym wykładnikiem są podwyższone stężenia troponin, zwłaszcza podczas wysiłku. U pacjentów z HFpEF i współistniejącą otyłością leczenie inkretynowe wykazuje korzystny wpływ na przerost lewej komory, stan zapalny i uszkodzenie mięśnia sercowego, natomiast hamowanie neprylizyny może dodatkowo wspierać ochronę funkcji nerek i spowalniać progresję niewydolności nerkowej.
Dostępne dane wskazują, że otyłość i HFpEF opierają się na tych samych mechanizmach patofizjologicznych, takich jak retencja sodu, aktywacja neurohormonalna, przewlekły stan zapalny, włóknienie narządów oraz nasilona sygnalizacja osi leptyna–aldosteron–neprylizyna. Kluczowe znaczenie ma jednak fakt, że otyłość trzewna i centralna nie tylko współistnieją z HFpEF, ale ją poprzedzają i przewidują jej rozwój, w przeciwieństwie do HFrEF. Badania eksperymentalne oraz obserwacje populacyjne konsekwentnie pokazują, że nadmiar tkanki tłuszczowej trzewnej jest wczesnym i silnym czynnikiem ryzyka rozwoju HFpEF, wykazując zależność typu dawka–odpowiedź, szczególnie w odniesieniu do tłuszczu otaczającego serce. W momencie rozpoznania HFpEF niemal wszyscy pacjenci wykazują nadmierną otyłość lub otyłość centralną, a nasilenie zaburzeń hemodynamicznych i klinicznych rośnie wraz ze stopniem otłuszczenia. Zbieżność mechanistyczna i czasowa przemawia za uznaniem otyłości trzewnej za jeden z głównych czynników napędzających patogenezę HFpEF. Zgodnie z hipotezą adipokinową, przerośnięta i zapalnie zmieniona tkanka tłuszczowa trzewna oddziałuje na serce, naczynia i nerki poprzez wydzielanie biologicznie aktywnych adipokin, sprzyjając rozwojowi dysfunkcji wielonarządowej. Dostępne dane nie potwierdzają, aby niedobór insuliny lub hiperinsulinemia bezpośrednio napędzały rozwój HFpEF.
Sygnalizacja międzynarządowa tkanki tłuszczowej i serca
W warunkach fizjologicznych tkanka tłuszczowa pełni funkcję ochronną, magazynując energię, detoksykując nadmiar kwasów tłuszczowych i hamując stan zapalny. W stanie nadmiaru składników odżywczych oraz w HFpEF dochodzi jednak do rozrostu trzewnej tkanki tłuszczowej, któremu towarzyszy stres adipocytów, przewlekły stan zapalny i procesy włóknienia. Zmiany te obejmują utratę zdolności regeneracyjnych komórek macierzystych oraz nasilone przekształcenie śródbłonka w mezenchymę.Dysfunkcyjna tkanka tłuszczowa wydziela liczne biologicznie aktywne cząsteczki, które podtrzymują lokalny stres oksydacyjny i zapalny, a po przedostaniu się do krążenia przenoszą te procesy do innych narządów, zwłaszcza serca. Adipocyty produkują setki białek i mediatorów sygnałowych, których profil w zdrowiu jest cytoprotekcyjny, natomiast w otyłości i HFpEF ulega przesunięciu w kierunku prozapalnym.Co istotne, te same szlaki wykrywania nadmiaru lub niedoboru składników odżywczych funkcjonują zarówno w tkance tłuszczowej, jak i w mięśniu sercowym. Ten paralelizm sygnalizacji wskazuje na istnienie ścisłej komunikacji międzynarządowej, która w warunkach patologicznych sprzyja rozwojowi i podtrzymywaniu HFpEF.
Brak równowagi sygnalizacji wrażliwej na składniki odżywcze
W zdrowym sercu dominują sygnały niedoboru składników odżywczych, które chronią kardiomiocyty poprzez wsparcie metabolizmu energetycznego, funkcji mitochondriów i autofagii (szlaki SIRT1–AMPK–PGC-1α). W niewydolności serca dochodzi do stanu pozornego nadmiaru składników odżywczych, z zahamowaniem autofagii, dysfunkcją mitochondriów i nasileniem stresu oksydacyjnego. Towarzyszy temu przewlekła aktywacja szlaków prohipertroficznych i profibrotycznych, głównie PI3K–Akt–mTOR, przy jednoczesnym osłabieniu sygnałów ochronnych. Podobny brak równowagi występuje w otyłości trzewnej, gdzie rozrośnięta tkanka tłuszczowa wykazuje dominację sygnału mTOR i zahamowanie AMPK/SIRT1. Zbieżność tych zmian w adipocytach i kardiomiocytach wskazuje na ścisłą komunikację międzynarządową, w której zaburzenia sygnalizacji w tkance tłuszczowej napędzają rozwój HFpEF.
Kluczowe dowody wspierające hipotezę adipokinową HFpEF
Zgromadzone dane jednoznacznie wskazują, że otyłość trzewna jest zdarzeniem poprzedzającym i niemal powszechną cechą HFpEF, a adipokiny stanowią główny mechanizm przekazywania dysfunkcyjnego „sygnału biologicznego” z tkanki tłuszczowej do serca.
1. Otyłość trzewna jako czynnik sprawczy HFpEF
Badania eksperymentalne i populacyjne pokazują, że nadmierna otyłość trzewna pojawia się na lata przed rozpoznaniem HFpEF i przewiduje jej rozwój, ale nie HFrEF. Otyłość centralna występuje u niemal wszystkich pacjentów z HFpEF, a stopień otłuszczenia koreluje z nasileniem zaburzeń hemodynamicznych, ciężkością objawów i rokowaniem.
2. Centralna rola adipokin
Adipocyty – w przeciwieństwie do kardiomiocytów – syntetyzują i wydzielają liczne kardioaktywne adipokiny. W otyłości trzewnej profil adipokin przesuwa się w stronę prozapalną i profibrotyczną. Zmiany stężeń adipokin:
-
pojawiają się przed HFpEF,
-
przewidują jej rozwój,
-
odzwierciedlają ciężkość choroby i rokowanie,
-
są zbieżne w otyłości i HFpEF.
Adipokiny mają udokumentowany wpływ na przerost, włóknienie, stres kardiomiocytów i dysfunkcję mikrokrążenia.
3. Korzyści z interwencji ukierunkowanych na tkankę tłuszczową
Operacje bariatryczne oraz leczenie farmakologiczne HFpEF prowadzą do nieproporcjonalnej redukcji tłuszczu trzewnego, czemu towarzyszy wzrost adipokin cytoprotekcyjnych i spadek prozapalnych. Wyjaśnia to, dlaczego pacjenci z otyłością częściej odnoszą korzyści z aktualnych terapii HFpEF. Badania eksperymentalne pokazują, że selektywna modulacja adipokin wyłącznie w tkance tłuszczowej (bez ingerencji w serce) wywołuje istotne zmiany fenotypu serca, potwierdzając istnienie osi tkanka tłuszczowa–serce.
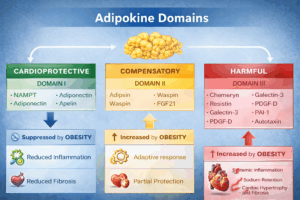
4. Funkcjonalna klasyfikacja adipokin
Zaproponowano nowy podział kardioaktywnych adipokin na trzy domeny:
-
Domena I – adipokiny ochronne, obecne u osób szczupłych; ich spadek w otyłości sprzyja HFpEF.
-
Domena II – adipokiny kompensacyjne, wzrastające w otyłości, lecz niewystarczające (często z powodu oporności).
-
Domena III – adipokiny prozapalne i profibrotyczne, dominujące w otyłości trzewnej i napędzające HFpEF.
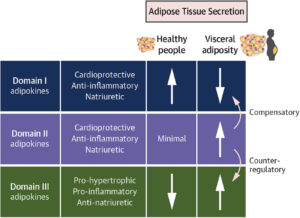
Zmiany w domenach I i III mają zwykle kierunek przeciwny, co dodatkowo wzmacnia patogenetyczny model adipokinowy HFpEF. Całość dowodów wskazuje, że HFpEF jest w dużej mierze chorobą napędzaną przez dysfunkcyjną tkankę tłuszczową trzewną, a adipokiny stanowią kluczowe ogniwo łączące otyłość z patologią serca.
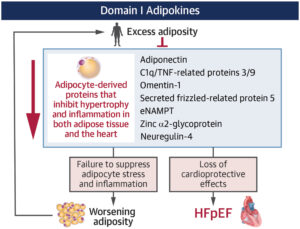
Adipokiny domeny I: kluczowe cząsteczki kardioprotekcyjne
Adipokiny domeny I są syntetyzowane głównie przez adipocyty osób szczupłych i pełnią funkcję ochronną wobec serca. Hamują przerost i włóknienie mięśnia sercowego, ograniczają stan zapalny oraz sprzyjają prawidłowej adaptacji metabolicznej, chroniąc przed rozwojem HFpEF. Ich działanie jest mediowane m.in. przez aktywację szlaków SIRT1–AMPK–PGC-1α, modulację sygnalizacji Wnt oraz receptory sprzężone z białkiem G. W otyłości trzewnej i HFpEF dochodzi do supresji tych adipokin, co usuwa istotny mechanizm kardioprotekcyjny. Najważniejszą adipokiną domeny I jest adiponektyna, której wysokie stężenia u osób szczupłych sprzyjają wrażliwości insulinowej, zdrowiu mitochondriów i hamowaniu stresu oksydacyjnego. Adiponektyna antagonizuje działanie aldosteronu i aktywność układu współczulnego. W otyłości trzewnej i HFpEF jej poziom jest obniżony, co wiąże się z retencją sodu, wzrostem objętości osocza, powiększeniem lewego przedsionka i przerostem lewej komory. Niskie stężenia adiponektyny przewidują gorsze rokowanie sercowo-naczyniowe. Do adipokin adiponektynopodobnych należą CTRP3 i CTRP9, które również aktywują AMPK i SIRT1 oraz wykazują działanie przeciwzapalne, wazodylatacyjne i kardioprotekcyjne. Ich stężenia są obniżone w otyłości trzewnej i niewydolności serca, a eksperymentalne uzupełnianie CTRP łagodzi uszkodzenie mięśnia sercowego, co wskazuje na dwukierunkową komunikację między tkanką tłuszczową a sercem.
Inne istotne adipokiny domeny I obejmują:
-
Omentynę-1 – działa przeciwzapalnie, wazodylatacyjnie i przeciwnadciśnieniowo; jej poziom jest obniżony w otyłości i HFpEF, a wzrasta po terapii agonistami GLP-1.
-
SFRP5 – antagonizuje prozapalną sygnalizację Wnt; niskie stężenia wiążą się z gorszą funkcją rozkurczową i rokowaniem w HF.
-
eNAMPT – kluczowy regulator biosyntezy NAD⁺ i aktywności SIRT1; jego niedobór w otyłości i HFpEF sprzyja stresowi oksydacyjnemu i dysfunkcji serca.
-
α2-glikoproteinę cynkową (ZAG) – wspiera lipolizę, natriurezę i działa przeciwwłóknieniowo; w HFpEF związanym z otyłością jej poziomy są obniżone.
-
Neuregulinę-4 (NRG-4) – adipokinę brunatnej tkanki tłuszczowej, która poprawia funkcję mitochondriów, zmniejsza zapalenie i łagodzi kardiomiopatię; jej ekspresja spada w otyłości trzewnej.
Adipokiny domeny I stanowią podstawowy system ochronny serca, który ulega zahamowaniu w otyłości trzewnej. Utrata ich działania usuwa barierę przed przerostem, zapaleniem i włóknieniem, tworząc warunki sprzyjające rozwojowi HFpEF.
Adipokiny domeny II
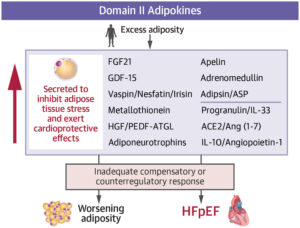
Adipokiny domeny II to cząsteczki o działaniu kardioprotekcyjnym, których wydzielanie rośnie w otyłości (zwłaszcza trzewnej) jako mechanizm kompensacyjny: mają częściowo zastąpić utraconą ochronę adipokin domeny I i równoważyć szkodliwe działanie adipokin domeny III. Hamują stres komórkowy, przerost, stan zapalny i włóknienie w tkance tłuszczowej oraz w sercu. U osób szczupłych ich stężenia są zwykle niskie (często kojarzone z wątrobą, mięśniami czy OUN), natomiast w otyłości adipocyty stają się głównym źródłem tych cząsteczek. Mimo wzrostu ich poziomów HFpEF nadal się rozwija — prawdopodobnie dlatego, że mechanizm kompensacyjny jest niewystarczający lub pojawia się oporność tkankowa na ich działanie. Wiele adipokin domeny II działa przez „ochronne” szlaki SIRT1/AMPK/PGC-1α oraz hamowanie osi PI3K–Akt–mTOR, zmniejszając skutki nadmiaru składników odżywczych.
Najważniejsze przykłady:
-
FGF21 – w otyłości rośnie jego produkcja w tkance tłuszczowej; poprawia metabolizm, sprzyja lipolizie i redukcji tłuszczu trzewnego, działa też kardioprotekcyjnie. Jednocześnie w otyłości/cukrzycy rozwija się oporność na FGF21, więc mimo wysokich stężeń efekt bywa ograniczony.
-
GDF-15 – hormon stresu metabolicznego: zmniejsza apetyt, wspiera lipolizę i działa przeciwzapalnie; może łagodzić przerost i włóknienie oraz zaburzenia rozkurczu. W otyłości często jest podwyższony, co może odzwierciedlać reakcję obronną (z możliwą opornością).
-
Waspina, nesfatyna-1, iryzyna – adipokiny kontrregulacyjne zwiększane w otyłości; wspierają AMPK/SIRT1, nasilają autofagię, ograniczają stan zapalny i mogą poprawiać funkcję serca. W HFpEF wyższe stężenia (np. iryzyny) bywają związane z lepszym rokowaniem.
-
Metalotioneina-1 – odpowiedź antyoksydacyjna adipocytów; zmniejsza stres oksydacyjny w sercu i może poprawiać funkcję rozkurczową.
-
HGF – wzrasta w otyłości i może zapowiadać rozwój HFpEF; mimo że jest traktowany jako czynnik ochronny, jego podwyższone stężenia są też markerem ryzyka i przebudowy serca.
-
Apelina oraz adrenomedulina/CGRP – peptydy działające przez receptory sprzężone z białkiem G; poprawiają funkcję naczyniową, działają inotropowo i przeciwprzerostowo, a w otyłości ich poziomy często rosną jako przeciwregulacja.
-
Adipsyna/ASP – wspiera „bezpieczne” magazynowanie tłuszczu i ogranicza ektopowe otłuszczenie trzewne; jej wzrost w otyłości może być mechanizmem ochronnym, a badania eksperymentalne wskazują na możliwy wpływ na fenotyp HFpEF.
-
Naturalni antagoniści adipokin domeny III (np. ACE2/Ang(1-7), IL-10, Ang-1, progranulina) – ograniczają zapalenie, włóknienie i dysfunkcję naczyniową, działając jako „hamulec” dla prozapalnych szlaków.
Adipokiny domeny II są próbą „ratunkowej” odpowiedzi organizmu na otyłość trzewną — jednak w HFpEF często okazują się za słabe lub blokowane przez oporność, dlatego choroba nadal postępuje.
Adipokiny domeny III i HFpEF
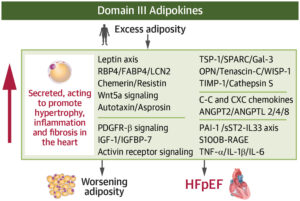
W otyłości, szczególnie trzewnej, przerośnięta i zapalna tkanka tłuszczowa zmienia swój profil wydzielniczy w kierunku adipokin domeny III, które wywierają bezpośrednio szkodliwy wpływ na serce i napędzają rozwój HFpEF. Są one produkowane głównie przez dysfunkcyjne adipocyty oraz komórki zapalne tkanki tłuszczowej i aktywują szlaki PI3K–Akt–mTOR, JAK/STAT, Wnt oraz TGF-β/Smad, prowadząc do przerostu, zapalenia, włóknienia i dysfunkcji rozkurczowej mięśnia sercowego.Do kluczowych mediatorów należą m.in. leptyna (oraz oś leptyna–RAA–neprylizyna), lipokaliny (RBP4, FABP4, LCN2), rezystyna, chemeryna, Wnt5a, autotaksyna, asprosyna, a także liczne czynniki wzrostu, chemokiny, angiopoetyny oraz białka macierzy zewnątrzkomórkowej (np. galektyna-3, trombospondyna-1, TIMP1).
Adipokiny te:
-
nasilają stan zapalny i insulinooporność,
-
powodują retencję sodu, dysfunkcję śródbłonka i mikrokrążenia,
-
indukują przerost i włóknienie serca,
-
korelują z nieprawidłowościami napełniania rozkurczowego, objawami HFpEF i gorszym rokowaniem.
Podwyższone ich stężenia we krwi mają znaczenie prognostyczne, a ich hamowanie (farmakologiczne lub po redukcji masy ciała) łagodzi zaburzenia metaboliczne i sercowe. Adipokiny domeny III stanowią więc centralne ogniwo patofizjologiczne łączące otyłość z HFpEF i potencjalny cel terapeutyczny.
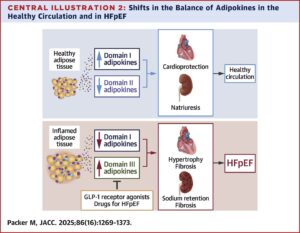
Testowanie hipotezy adipokinowej HFpEF i rola terapii
Hipoteza adipokinowa HFpEF zakłada, że ekspansja i dysfunkcja trzewnej tkanki tłuszczowej napędza HFpEF poprzez zaburzone wydzielanie bioaktywnych cząsteczek (adipokin), które uszkadzają serce, naczynia i nerki. Dlatego sensowny paradygmat musi tłumaczyć, czy obecne i przyszłe terapie HFpEF działają m.in. przez redukcję otyłości trzewnej i normalizację profilu adipokin.
Chirurgia bariatryczna – „modelowy dowód” hipotezy
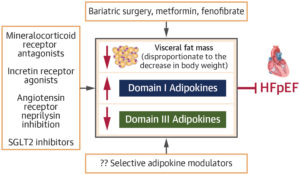
Najmocniejsze potwierdzenie hipotezy daje operacja bariatryczna (np. gastric bypass), bo wywołuje:
-
dużą utratę masy ciała z nieproporcjonalnie dużą redukcją tłuszczu trzewnego,
-
spadek zapalenia tkanki tłuszczowej (głównie przez wyciszenie genów prozapalnych w adipocytach, a nie przez bezpośredni wpływ na krążące monocyty).
„Przełączenie” profilu adipokin po operacji
Po bariatrii obserwuje się typowy wzorzec:
-
↑ adipokiny domeny I (zwłaszcza adiponektyna),
-
↓ adipokiny domeny III (prawie „globalnie”), w tym m.in.: leptyna i elementy osi RAA/neprylizyna, FABP4, RBP4, YKL-40, chemeryna, rezystyna, Wnt5a, asprosyna, aktywina A, trombospondyna-1, SPARC, TIMP1, katepsyna S, CCL2, ANGPTL2, PAI-1, sST2 oraz część cytokin prozapalnych (TNF-α, IL-1β, IL-6),
-
zwykle ↓ adipokiny domeny II (kompensacyjne/„równoważące”), bo maleje potrzeba ich kontr-regulacji,
To pokazuje, że sam deficyt energetyczny + poprawa biologii tkanki tłuszczowej potrafi przesunąć organizm do nowego „set-pointu” adipokin, potencjalnie korzystnego dla serca.
Po operacji bariatrycznej:
-
zmniejsza się przerost lewej komory,
-
poprawia się mikrokrążenie wieńcowe i funkcja rozkurczowa,
-
efekty te korelują ze spadkiem tłuszczu trzewnego i zmianami adipokin, a częściowo mogą być niezależne od samej utraty masy ciała,
-
klinicznie: spada ryzyko nowej niewydolności serca i szczególnie hospitalizacji z powodu HFpEF.
Wniosek: bariatryczna „reprogramacja” trzewnej tkanki tłuszczowej i jej sekretomu jest najbardziej spójnym, praktycznym przykładem, że adipokiny są prawdopodobnym mechanizmem łączącym otyłość trzewną z HFpEF i że skuteczne terapie powinny (bezpośrednio lub pośrednio) celować w VAT i profil adipokin.
Antagoniści receptora mineralokortykoidowego (MRA)
- Aldosteron jest adipokiną domeny III, a adipocyty są jego istotnym źródłem w otyłości trzewnej.
- Istnieje błędne koło: otyłość trzewna ↑ aldosteron, a aldosteron nasila dysfunkcję metaboliczną i sercowo-naczyniową.
- MRA (spironolakton, eplerenon):
- zmniejszają masę tłuszczu trzewnego i obwód talii (często niezależnie od całkowitej masy ciała),
- hamują stan zapalny tkanki tłuszczowej,
- zapobiegają przyrostowi masy ciała u pacjentów z HFpEF,
- poprawiają rozkurczowe napełnianie komór nawet w dawkach subantyhipertensyjnych.
Wpływ MRA na adipokiny
- ↑ adipokiny domeny I (np. adiponektyna, eNAMPT),
- ↑ adipokiny domeny II (m.in. GDF-15, adrenomedulina, apelina, HGF),
- ↓ adipokiny domeny III (leptyna, chemeryna, PAI).
Efektem jest przesunięcie równowagi adipokin w kierunku mniejszego stresu tkanki tłuszczowej i serca.
- Korzyści z MRA są największe u pacjentów z otyłością centralną.
- W HFpEF obserwuje się zależność między BMI a skutecznością leczenia: finerenon i spironolakton redukują ryzyko zdarzeń sercowo-naczyniowych głównie u pacjentów z otyłością.
Wniosek: skuteczność MRA w HFpEF można w dużej mierze wyjaśnić ich działaniem na trzewną tkankę tłuszczową i normalizację profilu adipokin, co stanowi silne kliniczne wsparcie dla hipotezy adipokinowej HFpEF.
Agoniści receptora GLP-1 i dualni agoniści GLP-1/GIP
-
Mechanizm: bezpośrednie działanie na adipocyty → ↓ przerost i zapalenie, ↑ funkcja mitochondriów i termogeneza BAT (SIRT1). Agonizm GIP nasila lipolizę i wzmacnia efekt redukcji masy.
-
Tkanka tłuszczowa: nieproporcjonalnie duża redukcja tłuszczu trzewnego, w tym nasierdziowego i przysercowego (większa niż spadek masy ciała).
-
Profil adipokin: ↑ domena I (adiponektyna, CTRP3, omentyna-1, SFRP5, eNAMPT, ZAG), ↓ domena III (m.in. leptyna, aldosteron, RBP4, FABP4, rezystyna, PAI, cytokiny prozapalne); częściowo ↑ domena II (np. FGF21, iryzyna).
-
Efekt kliniczny: poprawa objawów i tolerancji wysiłku w HFpEF z otyłością; największe korzyści u chorych z najwyższym BMI i największą utratą masy/trzewnej tkanki tłuszczowej. Zmniejszeniu EAT towarzyszy regresja przerostu LV i powiększenia LA.
Inhibitory SGLT2
-
Mechanizm: indukcja „mimikry głodu” (AMPK/SIRT1/PGC-1α), ↑ autofagia; działanie niezależne od glikozurii także w adipocytach.
-
Tkanka tłuszczowa: ↓ hipertrofia i zapalenie adipocytów, selektywna redukcja tłuszczu trzewnego i EAT przy niewielkich zmianach masy ciała (kompensacyjna hiperfagia).
-
Profil adipokin: ↑ domena I (adiponektyna, ZAG) i domena II (FGF21, GDF-15, apelina); ↓ domena III (leptyna, RBP4, chemeryna, asprozyna, PAI, CCL2, TNF-α, IL-6).
-
Efekt kliniczny: poprawa hemodynamiki (↓ ciśnienie napełniania LV), większe korzyści u pacjentów z otyłością, zwłaszcza z T2D; cytoprotekcja kardiomiocytów pośredniczona przez „odprogramowane” adipocyty.
Wniosek: Zarówno terapie inkretynowe, jak i inhibitory SGLT2 wspierają hipotezę adipokinową HFpEF: redukują trzewną tkankę tłuszczową i przesuwają równowagę adipokin w kierunku kardioprotekcji, a skala korzyści zależy od wyjściowego nasilenia otyłości trzewnej i jej redukcji w trakcie leczenia.
Hamowanie receptora angiotensyny i neprylizyny w HFpEF
-
Angiotensyna II i neprylizyna są adipokinami domeny III, a ich nadmierna aktywacja w HFpEF jest w dużej mierze napędzana przez otyłość trzewną. Adipocyty bezpośrednio je wydzielają oraz pośrednio nasilają ich produkcję poprzez aktywację układu współczulnego (np. przez leptynę).
-
Redukcja tłuszczu trzewnego (np. po operacji bariatrycznej) prowadzi do spadku poziomów angiotensyny II i neprylizyny, co potwierdza przyczynową rolę tkanki tłuszczowej w aktywacji układów neurohormonalnych.
-
Blokada receptora angiotensyny (ARB):
-
zmniejsza masę tłuszczu trzewnego (nieproporcjonalnie do spadku masy ciała),
-
ogranicza przerost, stan zapalny i stres adipocytów,
-
zwiększa adipokiny kardioprotekcyjne (domena I i II, np. adiponektyna, apelina),
-
obniża adipokiny prozapalne (domena III).
-
-
Hamowanie neprylizyny dodatkowo:
-
nasila sygnalizację peptydów natriuretycznych,
-
hamuje adipogenezę, zwiększa lipolizę i termogenezę brunatnej tkanki tłuszczowej,
-
działa jak „przełącznik adipokin”, przesuwając profil wydzielniczy w stronę kardioprotekcji,
-
zwiększa stężenia korzystnych adipokin domeny II (adrenomedulina, apelina, Ang(1–7)).
-
-
Sakubitryl/walsartan:
-
zmniejsza masę tłuszczu trzewnego,
-
poprawia profil adipokinowy,
-
obniża markery włóknienia i zapalenia (sST2, TIMP1, YKL-40),
-
jest najskuteczniejszy u pacjentów z HFpEF i otyłością centralną.
-
-
Korzyści kliniczne ARNI w HFpEF są największe u chorych z niskimi poziomami peptydów natriuretycznych przed leczeniem, co pośrednio wskazuje na otyłość trzewną jako kluczowy czynnik odpowiedzi.
Wniosek:
Skuteczność hamowania receptora angiotensyny i neprylizyny w HFpEF wynika w dużej mierze z normalizacji biologii trzewnej tkanki tłuszczowej i profilu adipokin, a nie wyłącznie z klasycznych efektów hemodynamicznych.
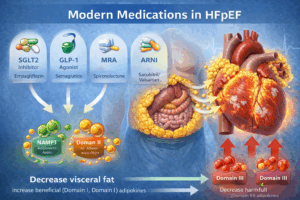
Perspektywy, synteza i repurposing leków w HFpEF
Perspektywa i synteza
-
MRA, agoniści inkretynowi, inhibitory SGLT2 i ARNI korzystnie modulują biologię adipocytów, prowadząc do nieproporcjonalnej redukcji tłuszczu trzewnego (często większej niż spadek masy ciała).
-
Towarzyszy temu wzrost adipokin domeny I, spadek adipokin domeny III oraz względny wzrost adipokin domeny II – przesunięcie profilu w stronę kardioprotekcji.
-
W HFpEF klasyczne bodźce hemodynamiczne są słabsze niż w HFrEF; otyłość trzewna staje się głównym źródłem aldosteronu, angiotensyny II i neprylizyny, co tłumaczy lepszą odpowiedź pacjentów z otyłością na leczenie neurohormonalne.
-
Redukcja tłuszczu trzewnego (np. po bariatrii) normalizuje aktywację neurohormonalną.
Możliwość ponownego wykorzystania leków (repurposing)
Metformina
-
Aktywator AMPK; hamuje adipogenezę, stan zapalny i włóknienie tkanki tłuszczowej; poprawia funkcję brunatnej tkanki tłuszczowej.
-
Zmniejsza tłuszcz trzewny i nasierdziowy, poprawia rozkurcz i sztywność LK; ogranicza rozwój HFpEF (zwłaszcza otyłościowej).
-
Zwiększa adipokiny domeny I, tłumi domenę III; wzorzec zmian podobny do bariatrii.
-
Korzyści kliniczne szczególnie u pacjentów z nadwagą/otyłością; u chorych z HFpEF – niższa śmiertelność.
Fenofibrat vs. pioglitazon
-
PPARα (fenofibrat): ↓ przerost i zapalenie adipocytów, ↑ termogeneza BAT, ↓ tłuszcz trzewny; kardioprotekcja; klinicznie ↓ hospitalizacji HF.
-
PPARγ (pioglitazon): ↑ adipogeneza, ↑ masa ciała i otyłość centralna, retencja sodu; zwiększone ryzyko HF.
-
Kluczowy jest kierunek sygnalizacji PPARα vs PPARγ – korzystny dla serca i tkanki tłuszczowej jest PPARα.
Wniosek
-
HFpEF to w dużej mierze choroba tkanki tłuszczowej trzewnej.
-
Leki, które zmniejszają jej masę i normalizują profil adipokin, oferują największe korzyści – zwłaszcza u pacjentów z otyłością centralną.
-
Repurposing (metformina, fenofibrat) ma silne podstawy biologiczne i kliniczne w HFpEF.
Inhibitory TNF-α, antagoniści IL-1/IL-1β i kolchicyna w HFpEF
Leki blokujące klasyczne cytokiny prozapalne (TNF-α, IL-1/IL-1β) oraz kolchicyna skutecznie zmniejszają stan zapalny, jednak nie przynoszą korzyści klinicznych w HFpEF związanej z otyłością.Kluczowe wnioski:
- W badaniach klinicznych antagoniści TNF-α (etanercept, infliksimab):
- nie poprawiają profilu adipokin,
- sprzyjają przyrostowi masy ciała i otyłości trzewnej,
- otyłość osłabia ich skuteczność przeciwzapalną.
- Antagoniści IL-1 (anakinra, kanakinumab):
- nie poprawiają funkcji ani wydolności u pacjentów z HFpEF i otyłością,
- nie wpływają korzystnie na leptynę ani adiponektynę,
- otyłość może neutralizować ich działanie.
- Kolchicyna:
- nie poprawia objawów ani rokowania w HFpEF,
- może hamować lipolizę i nie poprawia wrażliwości insulinowej.
- Łącznie: kanoniczne cytokiny prozapalne nie wydają się kluczowym napędem HFpEF związanej z otyłością, w przeciwieństwie do zaburzeń adipokinowych.
Perspektywa:
- Nowym kierunkiem jest blokada IL-6 – ziltivekimab jest obecnie oceniany w dużym badaniu klinicznym u pacjentów z HFpEF, szczególnie z otyłością i PChN.
- W badaniach klinicznych antagoniści TNF-α (etanercept, infliksimab):
Selektywne ukierunkowanie adipokin w HFpEF
Hipoteza adipokinowa wskazuje, że zaburzona równowaga adipokin domen I–III napędza patogenezę HFpEF, a jej celowana modulacja może stanowić nową strategię leczenia.
Najważniejsze tezy:
-
Wiele terapii ukierunkowanych na adipokiny jest już rozwijanych (głównie w otyłości, NAFLD/NASH, chorobach zapalnych i onkologii), ale nie testowano ich szeroko w HFpEF.
-
Obiecujące kierunki obejmują:
-
Wzmocnienie adipokin ochronnych (np. agoniści adiponektyny, FGF21, apeliny).
-
Hamowanie adipokin szkodliwych (np. leptyny, FABP4, YKL-40, galektyny-3, chemeryny, Wnt5a, RAGE).
-
Potencjalizację osi peptydów natriuretycznych (np. inhibitory PDE9).
-
Modulację szlaków zapalnych i włóknienia (np. sotatercept/aktywinA, pirfenidon).
-
-
Część leków wykazuje kardioprotekcję w modelach HFpEF, ale brakuje badań klinicznych w tej populacji.
-
Nie wszystkie strategie odchudzające są bezpieczne kardiologicznie (np. agonizm amyliny – ryzyko kardiotoksyczności).
-
Rozwój leków dla HFpEF jest spowalniany przez niską opłacalność rynkową i preferencję krótkoterminowych punktów końcowych.
Wniosek:
Lepsze zrozumienie patogenezy HFpEF uzasadnia repurposing i rozwój terapii ukierunkowanych na adipokiny, zwłaszcza tych stosowanych w otyłości trzewnej i NAFLD — schorzeniach ściśle powiązanych z HFpEF.
Tkanka tłuszczowa trzewna jest największym narządem wydzielniczym u osób z otyłością i kluczowym regulatorem sygnałów metabolicznych. W otyłości trzewnej dochodzi do przebudowy sekretomu adipocytów, z utraty profilu kardioprotekcyjnego na rzecz adipokin prozapalnych, prohipertroficznych i profibrotycznych, działających endokrynnie i parakrynnie na serce, nerki i naczynia.
Serce jest szczególnie wrażliwym celem, ponieważ:
-
jest narażone na zaburzone krążące adipokiny,
-
jest bezpośrednio otoczone nasierdziową tkanką tłuszczową, która w otyłości przekazuje sygnały zapalne i włókniejące,
-
jednocześnie wzrost objętości osocza (indukowany adipokinami) nasila obciążenie hemodynamiczne.
Kluczowe zagadnienia
HFpEF jest następstwem otyłości trzewnej, a nie jedynie jej współistniejącą chorobą. Zaburzone wydzielanie adipokin stanowi pierwotny mechanizm patogenetyczny HFpEF.
Adipokiny – nowa klasyfikacja
-
Domena I – adipokiny kardioprotekcyjne (↓ w otyłości)
-
Domena II – adipokiny kompensacyjne (↑ w otyłości, ale niewystarczająco)
-
Domena III – adipokiny prozapalne, retencyjne, profibrotyczne (↑ w otyłości)
Zmiany w domenach I i III są odwrotnie skorelowane i wykazują silny paralelizm w otyłości i HFpEF.
12 kluczowych linii dowodowych
-
Równoległa epidemia otyłości i HFpEF
-
Nadmiar kalorii jako zdarzenie inicjujące HFpEF
-
Otyłość i zmiany adipokin poprzedzają HFpEF
-
Prawie uniwersalna otyłość centralna w HFpEF
-
Silne nakładanie się cech biologicznych i klinicznych
-
Adipocyty jako główne źródło adipokin w otyłości
-
Równoległe zmiany adipokin z ciężkością HFpEF
-
Udokumentowany bezpośredni wpływ adipokin na serce
-
Chirurgia bariatryczna normalizuje adipokiny i HFpEF
-
Leki HFpEF zmniejszają tłuszcz trzewny i poprawiają profil adipokin
-
Otyłość identyfikuje pacjentów najlepiej odpowiadających na leczenie
-
Selektywna modulacja adipokin w tkance tłuszczowej zmienia strukturę i funkcję serca
Wniosek końcowy: HFpEF należy postrzegać jako chorobę adipokinową, napędzaną przez otyłość trzewną. Tkanka tłuszczowa jest centralnym regulatorem patogenezy HFpEF, a przyszłe strategie terapeutyczne powinny koncentrować się na redukcji tłuszczu trzewnego i normalizacji sygnalizacji adipokin – nie tylko na samym sercu.
Hipoteza adipokinowa HFpEF — synteza dowodów
1. Tkanka tłuszczowa jako nadrzędny regulator ogólnoustrojowy
U osób z otyłością tkanka tłuszczowa (często ≈50% masy ciała) staje się największym narządem wydzielniczym organizmu. Sekretom adipocytów przekształca bodźce żywieniowe w sygnały biologiczne (adipokiny), które działają endokrynnie, parakrynnie i juxtakrynnie na serce, nerki i naczynia. Ekspansja trzewnej tkanki tłuszczowej prowadzi do jej stresu, zapalenia i fundamentalnej zmiany profilu wydzielniczego – od cytoprotekcyjnego do prozapalnego, prohipertroficznego i profibrotycznego.
2. Serce jako szczególnie wrażliwy cel adipokin
Mięsień serca jest:
-
eksponowany na zmieniony profil krążących adipokin,
-
bezpośrednio otoczony przez biologicznie aktywną tkankę tłuszczową nasierdziową, która w otyłości wydziela adipokiny bezpośrednio do mięśnia sercowego.
Zdrowy tłuszcz nasierdziowy działa ochronnie, natomiast w otyłości przekazuje sygnały indukujące:
-
zapalenie,
-
przerost,
-
włóknienie,
-
dysfunkcję mikrokrążenia wieńcowego.
3. Trzy domeny adipokin — spójny model patogenetyczny HFpEF
Ekspansja trzewnej tkanki tłuszczowej powoduje zsynchronizowaną transformację sygnalizacji adipokinowej:
-
Domena I – adipokiny kardioprotekcyjne
↓ wydzielania w otyłości → utrata sygnałów adaptacyjnych -
Domena II – adipokiny kompensacyjne
↑ wydzielania w otyłości → reakcja obronna, często biologicznie niewystarczająca lub z opornością receptorową -
Domena III – adipokiny szkodliwe
↑ wydzielania → zapalenie ogólnoustrojowe, retencja sodu, przerost i włóknienie serca
Adipokiny z tej samej domeny zmieniają się równolegle, a domeny I i III wykazują trwałą zależność odwrotną — zarówno u osób zdrowych, jak i w HFpEF.
4. Dlaczego HFpEF nie jest klasyczną chorobą heterogenną
Chociaż HFpEF współwystępuje z licznymi chorobami (nadciśnienie, cukrzyca, CKD, AF, otyłość, choroby płuc), dane kliniczne nie potwierdzają istnienia wielu niezależnych ścieżek patogenetycznych.
Hipoteza adipokinowa proponuje, że: choroby współistniejące nie powodują HFpEF, HFpEF oraz jego powikłania są równoległymi manifestacjami jednego mechanizmu: dysfunkcyjnej trzewnej tkanki tłuszczowej. Analogicznie do HFrEF, gdzie różne objawy są skutkiem wspólnej aktywacji neurohormonalnej, w HFpEF adipokiny pełnią rolę nadrzędnych mediatorów.
5. Najsilniejsze dowody przyczynowe
Interwencje ukierunkowane wyłącznie na tkankę tłuszczową wykazują bezpośredni wpływ na serce i naczynia:
-
supresja adipokin domeny I (np. eNAMPT) → wielonarządowa dysfunkcja metaboliczna,
-
nadekspresja adipokin domeny II (np. adypsyna, waspina) → poprawa fenotypu HFpEF,
-
supresja adipokin domeny III (np. PDGF-D, rezystyna, autotaksyna, PAI-1) → zahamowanie przerostu i włóknienia serca.
Ponadto przeszczep tkanki tłuszczowej lub komórek szpiku od myszy z HFpEF przenosi fenotyp choroby, co stanowi jeden z najmocniejszych dowodów przyczynowości.
6. Kliniczne potwierdzenie hipotezy
Najbardziej przekonujące dane kliniczne pochodzą z:
-
chirurgii bariatrycznej – nieproporcjonalna redukcja tłuszczu trzewnego, normalizacja profilu adipokin, poprawa struktury i funkcji serca, redukcja hospitalizacji z powodu HFpEF,
-
leków skutecznych w HFpEF (MRA, inhibitory SGLT2, ARNI, agoniści inkretynowi) – wspólny mechanizm: redukcja trzewnej tkanki tłuszczowej + przesunięcie równowagi adipokin (↑ domena I, ↓ domena III).
Co istotne, skala korzyści klinicznych rośnie wraz z nasileniem otyłości trzewnej, co dodatkowo wspiera model adipokinowy.
7. Konsekwencje terapeutyczne
-
Celem leczenia HFpEF nie powinna być sama redukcja masy ciała, lecz przywrócenie prawidłowej biologii tkanki tłuszczowej.
-
Model wskazuje na racjonalność:
-
repurposing (metformina, fenofibrat),
-
terapii ukierunkowanych na adipokiny (FGF21, aktywina, PDE9, leptyna),
-
ostrożności wobec interwencji odchudzających o potencjalnej kardiotoksyczności.
-
8. Podsumowanie
Hipoteza adipokinowa HFpEF stanowi spójny model patogenetyczny, który:
-
integruje epidemiologię, patofizjologię, eksperymenty molekularne i badania kliniczne,
-
wyjaśnia związek otyłości trzewnej z HFpEF,
-
porządkuje pozorną heterogeniczność choroby,
-
tworzy logiczne podstawy dla nowych strategii terapeutycznych.